Explore Human BioMolecular Atlas Program Data Portal
Christine Hou
Department of Biostatistics, Johns Hopkins Universitychris2018hou@gmail.com Source:
vignettes/hubmapr_vignettes.Rmd
hubmapr_vignettes.RmdOverview
‘HuBMAP’ data portal (https://portal.hubmapconsortium.org/) provides an open,
global bio-molecular atlas of the human body at the cellular level.
HuBMAPR package provides an alternative interface to
explore the data via R.
The HuBMAP Consortium offers several APIs. To achieve
the main objectives, HuBMAPR package specifically
integrates three APIs:
Search API: The Search API is primarily searching relevant data information and is referenced to the Elasticsearch API.
Entity API: The Entity API is specifically utilized in the
bulk_data_transfer()function for Globus URL retrievalOntology API: The Ontology API is applied in the
organ()function to provide additional information about the abbreviation and corresponding full name of each organ.
Each API serves a distinct purpose with unique query capabilities,
tailored to meet various needs. Utilizing the httr2 and
rjsoncons packages, HuBMAPR effectively
manages, modifies, and executes multiple requests via these APIs,
presenting responses in formats such as tibble or character. These
outputs are further modified for clarity in the final results from the
HuBMAPR functions, and these functions help reflect the
data information of HuBMAP Data Portal as much as possible.
Using temporary storage to cache API responses facilitates efficient
data retrieval by reducing the need for redundant requests to the HuBMAP
Data Portal. This approach minimizes server load, improves response
times (e.g. datasets() takes less than 4 seconds to
retrieve more than 3500 records’ information, shown below), and enhances
overall query efficiency. By periodically clearing cached data or
directing them to a temporary directory, the process ensures that the
retrieved information remains relevant while managing storage
effectively. This caching mechanism supports a smoother and more
efficient user experience when accessing data from the portal.
HuBMAP Data incorporates three different identifiers:
HuBMAP ID, e.g. HBM399.VCTL.353
Universally Unique Identifier (UUID), e.g. 7036a70229eff1a51af965454dddbe7d
Digital Object Identifiers (DOI), e.g. 10.35079/HBM399.VCTL.353.
The HuBMAPR package utilizes the UUID - a 32-digit
hexadecimal number - and the more human-readable HuBMAP ID as two common
identifiers in the retrieved results. Considering precision and
compatibility with software implementation and data storage, UUID serves
as the primary identifier to retrieve data across various functions,
with the UUID mapping uniquely to its corresponding HuBMAP ID.
The systematic nomenclature is adopted for functions in the package by appending the entity category prefix to the concise description of the specific functionality. Most of the functions are grouped by entity categories, thereby simplifying the process of selecting the appropriate functions to retrieve the desired information associated with the given UUID from the specific entity category. The structure of these functions is heavily consistent across all entity categories with some exceptions for collection and publication.
Installation
HuBMAPR is a R package. The package can be installed
by
if (!requireNamespace("BiocManager")) {
install.packages("BiocManager")
}
BiocManager::install("HuBMAPR")Install development version from GitHub:
remotes::install_github("christinehou11/HuBMAPR")Basic User Guide
Implementation Notes
This session is to guide on extending or customizing the
HuBMAPR package to accommodate potential future changes in
data structure, enhancing the package’s long-term utility. We included a
brief outline to illustrate the basics of the principles and approach to
package design.
Identify an API endpoint
Provide an R client to translate R data structures to the arguments and parameters required by the API
Handle the response consistently to argument and response validation
Format the return value as a ‘tibble’ or ‘character’ to minimize cognitive demands on the user to interpret the result, and facilitate the incorporation into general R workflows
Load Necessary Packages
Load additional packages. dplyr package is widely used
in this vignette to conduct data wrangling and specific information
extraction.
Data Discovery
HuBMAP data portal page displays chronologically (last
modified date time) five categories of entity data:
Dataset
Sample
Donor
Publication
Collection.
Using corresponding functions to explore entity data.
system.time({
datasets_df <- datasets()
})
#> user system elapsed
#> 1.097 0.042 3.272
object_size(datasets_df)
#> 1.54 MB
datasets_df
#> # A tibble: 5,493 × 14
#> uuid hubmap_id dataset_type dataset_type_additio…¹ organ analyte_class
#> <chr> <chr> <chr> <chr> <chr> <chr>
#> 1 6e5d5a3cbc… HBM986.Q… CyTOF "" Blood Protein
#> 2 63005d98f9… HBM938.W… CyTOF "" Bone… Protein
#> 3 e137385cfe… HBM444.R… CyTOF "" Bone… Protein
#> 4 c585d35f67… HBM696.B… CyTOF "" Bone… Protein
#> 5 9465cf8e0f… HBM422.W… CyTOF "" Bone… Protein
#> 6 335c871203… HBM576.V… CyTOF "" Bone… Protein
#> 7 630144bf74… HBM547.N… CyTOF "" NA Protein
#> 8 a211b3570e… HBM843.Q… CyTOF "" Blood Protein
#> 9 607baa57ec… HBM628.F… CyTOF "" Blood Protein
#> 10 2825809b6a… HBM727.D… CyTOF "" Blood Protein
#> # ℹ 5,483 more rows
#> # ℹ abbreviated name: ¹dataset_type_additional_information
#> # ℹ 8 more variables: sample_category <chr>, status <chr>,
#> # dataset_processing_category <chr>, pipeline <chr>, registered_by <chr>,
#> # donor_hubmap_id <chr>, group_name <chr>, last_modified_timestamp <chr>samples(), donors(),
collections(), and publications() work same as
above.
The default tibble produced by the corresponding entity function only
reflects selected information. To see the names of selected information,
use the following commands for each entity category. Specify the
parameter of as to display information in the format of
"character" or "tibble".
# as = "tibble" (default)
datasets_col_tbl <- datasets_default_columns(as = "tibble")
datasets_col_tbl
#> # A tibble: 14 × 1
#> columns
#> <chr>
#> 1 uuid
#> 2 hubmap_id
#> 3 group_name
#> 4 dataset_type_additional_information
#> 5 dataset_type
#> 6 organ
#> 7 analyte_class
#> 8 dataset_processing_category
#> 9 sample_category
#> 10 registered_by
#> 11 status
#> 12 pipeline
#> 13 last_modified_timestamp
#> 14 donor_hubmap_id
# as = "character"
datasets_col_char <- datasets_default_columns(as = "character")
datasets_col_char
#> [1] "uuid" "hubmap_id"
#> [3] "group_name" "dataset_type_additional_information"
#> [5] "dataset_type" "organ"
#> [7] "analyte_class" "dataset_processing_category"
#> [9] "sample_category" "registered_by"
#> [11] "status" "pipeline"
#> [13] "last_modified_timestamp" "donor_hubmap_id"samples_default_columns(),
donors_default_columns(),
collections_default_columns(), and
publications_default_columns() work same as above.
A brief overview of selected information for five entity categories is:
tbl <- bind_cols(
dataset = datasets_default_columns(as = "character"),
sample = c(samples_default_columns(as = "character"), rep(NA, 7)),
donor = c(donors_default_columns(as = "character"), rep(NA, 6)),
collection = c(collections_default_columns(as = "character"),
rep(NA, 10)),
publication = c(publications_default_columns(as = "character"),
rep(NA, 7))
)
tbl
#> # A tibble: 14 × 5
#> dataset sample donor collection publication
#> <chr> <chr> <chr> <chr> <chr>
#> 1 uuid uuid hubm… uuid uuid
#> 2 hubmap_id hubmap_id uuid hubmap_id hubmap_id
#> 3 group_name group_name grou… title title
#> 4 dataset_type_additional_information sample_cate… Sex last_modi… publicatio…
#> 5 dataset_type organ Age NA last_modif…
#> 6 organ last_modifi… Body… NA publicatio…
#> 7 analyte_class donor_hubma… Race NA publicatio…
#> 8 dataset_processing_category NA last… NA NA
#> 9 sample_category NA NA NA NA
#> 10 registered_by NA NA NA NA
#> 11 status NA NA NA NA
#> 12 pipeline NA NA NA NA
#> 13 last_modified_timestamp NA NA NA NA
#> 14 donor_hubmap_id NA NA NA NAUse organ() to read through the available organs
included in HuBMAP. It can be helpful to filter retrieved
data based on organ information.
organs <- organ()
organs
#> # A tibble: 43 × 2
#> abbreviation name
#> <chr> <chr>
#> 1 BD Blood
#> 2 BL Bladder
#> 3 BM Bone Marrow
#> 4 BR Brain
#> 5 BV Blood Vasculature
#> 6 HT Heart
#> 7 LA Larynx
#> 8 LB Bronchus (Left)
#> 9 LE Eye (Left)
#> 10 LF Fallopian Tube (Left)
#> # ℹ 33 more rowsData Wrangling Examples
Data wrangling and filter are welcome to retrieve data based on interested information.
# Example from datasets()
datasets_df |>
filter(organ == 'Small Intestine') |>
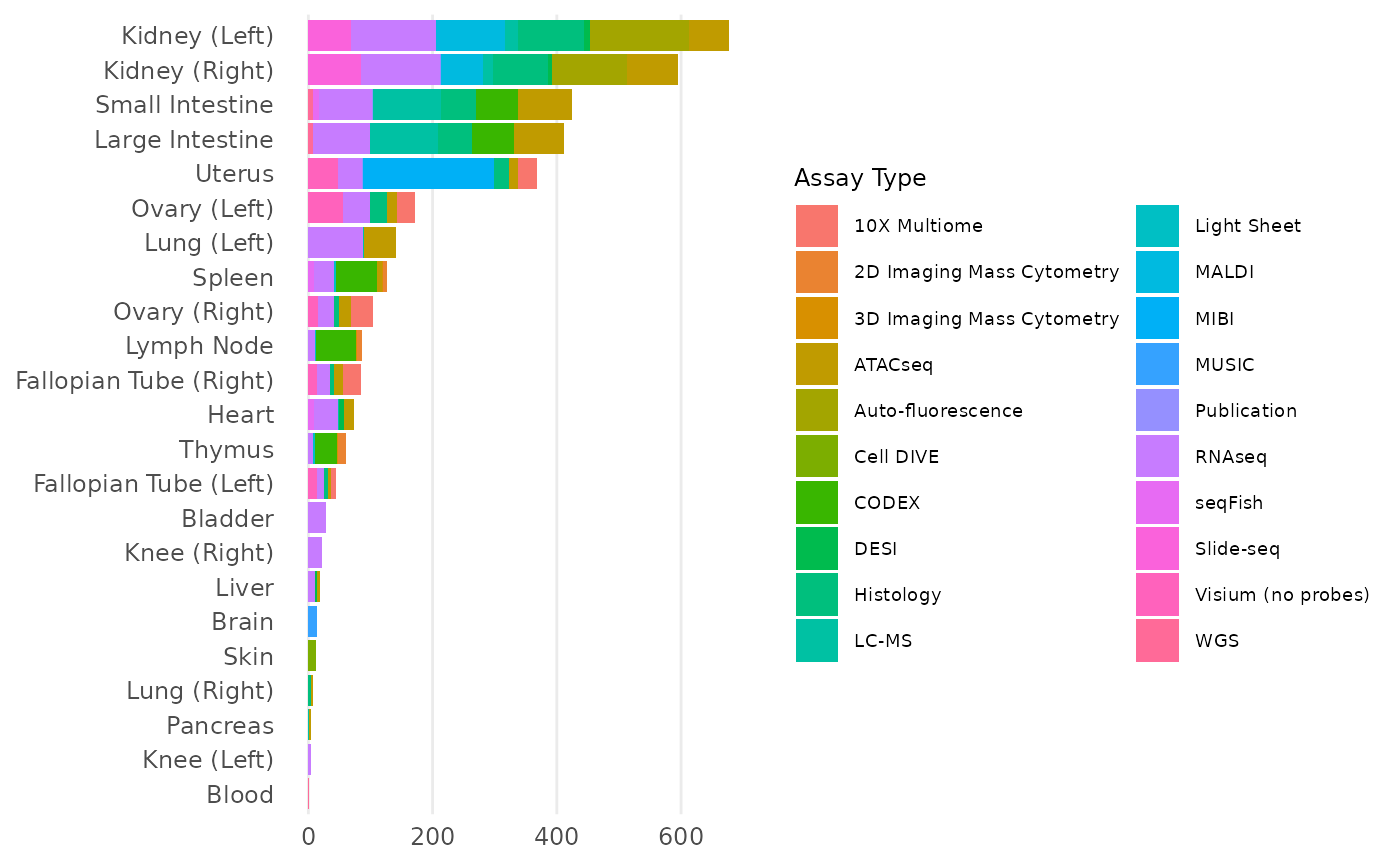
count()
#> # A tibble: 1 × 1
#> n
#> <int>
#> 1 423Any dataset, sample, donor, collection, and publication has a special HuBMAP ID and UUID, and UUID is the main ID to be used in most functions for specific detail retrievals.
The column of donor_hubmap_id is included in the
retrieved tibbles from samples() and
datasets(), which can help to join the tibble.
donors_df <- donors()
donor_sub <- donors_df |>
filter(Sex == "Female",
Age <= 76 & Age >= 55,
Race == "White",
`Body Mass Index` <= 25,
last_modified_timestamp >= "2020-01-08" &
last_modified_timestamp <= "2020-06-30") |>
head(1)
# Datasets
donor_sub_dataset <- donor_sub |>
left_join(datasets_df |>
select(-c(group_name, last_modified_timestamp)) |>
rename("dataset_uuid" = "uuid",
"dataset_hubmap_id" = "hubmap_id"),
by = c("hubmap_id" = "donor_hubmap_id"))
donor_sub_dataset
#> # A tibble: 0 × 19
#> # ℹ 19 variables: uuid <chr>, hubmap_id <chr>, group_name <chr>, Sex <chr>,
#> # Age <dbl>, Body Mass Index <dbl>, Race <chr>,
#> # last_modified_timestamp <chr>, dataset_uuid <chr>, dataset_hubmap_id <chr>,
#> # dataset_type <chr>, dataset_type_additional_information <chr>, organ <chr>,
#> # analyte_class <chr>, sample_category <chr>, status <chr>,
#> # dataset_processing_category <chr>, pipeline <chr>, registered_by <chr>
# Samples
samples_df <- samples()
donor_sub_sample <- donor_sub |>
left_join(samples_df |>
select(-c(group_name, last_modified_timestamp)) |>
rename("sample_uuid" = "uuid",
"sample_hubmap_id" = "hubmap_id"),
by = c("hubmap_id" = "donor_hubmap_id"))
donor_sub_sample
#> # A tibble: 0 × 12
#> # ℹ 12 variables: uuid <chr>, hubmap_id <chr>, group_name <chr>, Sex <chr>,
#> # Age <dbl>, Body Mass Index <dbl>, Race <chr>,
#> # last_modified_timestamp <chr>, sample_uuid <chr>, sample_hubmap_id <chr>,
#> # sample_category <chr>, organ <chr>You can use *_detail(uuid) to retrieve all available
information for any entry of any entity category given
UUID. Use select() and
unnest_*() functions to expand list-columns. It will be
convenient to view tables with multiple columns but one row using
glimpse().
dataset_uuid <- datasets_df |>
filter(dataset_type == "Auto-fluorescence",
organ == "Kidney (Right)") |>
head(1) |>
pull(uuid)
# Full Information
dataset_detail(dataset_uuid) |> glimpse()
#> Rows: 1
#> Columns: 30
#> $ ancestor_ids <list> <"ac5fc992bcb4c8234bb6f8c4ff751159",…
#> $ ancestors <list> [["Auto-fluorescence", "ac5fc992bcb4…
#> $ contains_human_genetic_sequences <lgl> FALSE
#> $ created_by_user_displayname <chr> "HuBMAP Process"
#> $ created_by_user_email <chr> "hubmap@hubmapconsortium.org"
#> $ created_timestamp <dbl> 1.744682e+12
#> $ creation_action <chr> "Central Process"
#> $ data_access_level <chr> "public"
#> $ dataset_info <chr> "ometiff_pyramid__ac5fc992bcb4c8234bb…
#> $ dataset_type <chr> "Auto-fluorescence [Image Pyramid]"
#> $ descendant_ids <list> []
#> $ descendants <list> []
#> $ display_subtype <chr> "Auto-fluorescence [Image Pyramid]"
#> $ donor <list> ["Jamie Allen", "jamie.l.allen@vander…
#> $ entity_type <chr> "Dataset"
#> $ files <list> [["OME-TIFF pyramid file", "EDAM_1.24…
#> $ group_name <chr> "Vanderbilt TMC"
#> $ group_uuid <chr> "73bb26e4-ed43-11e8-8f19-0a7c1eab007a"
#> $ hubmap_id <chr> "HBM232.PVNN.929"
#> $ immediate_ancestor_ids <list> "ac5fc992bcb4c8234bb6f8c4ff751159"
#> $ immediate_descendant_ids <list> []
#> $ index_version <chr> "3.6.3"
#> $ ingest_metadata <list> [[[<NULL>, "39be8df7", [], "https://g…
#> $ last_modified_timestamp <dbl> 1.746803e+12
#> $ origin_samples <list> [["Jamie Allen", "jamie.l.allen@vande…
#> $ published_timestamp <dbl> 1.746803e+12
#> $ source_samples <list> [["Jamie Allen", "jamie.l.allen@vande…
#> $ status <chr> "Published"
#> $ title <chr> "Auto-fluorescence [Image Pyramid] da…
#> $ uuid <chr> "262d079257f3932597bc99e5b1d7a362"
# Specific Information
dataset_detail(uuid = dataset_uuid) |>
select(title)
#> # A tibble: 1 × 1
#> title
#> <chr>
#> 1 Auto-fluorescence [Image Pyramid] data from the kidney (right) of a 57-year-o…sample_detail(), donor_detail(),
collection_detail(), and publication_detail()
work same as above.
Metadata
To retrieve the metadata for Dataset,
Sample, and Donor metadata, use
dataset_metadata(), sample_metadata(), and
donor_metadata().
dataset_metadata("993bb1d6fa02e2755fd69613bb9d6e08")
#> New names:
#> • `` -> `...1`
#> # A tibble: 20 × 2
#> Key Value
#> <chr> <chr>
#> 1 acquisition_instrument_model "Axio Scan.Z1"
#> 2 acquisition_instrument_vendor "Zeiss Microscopy"
#> 3 analyte_class "Endogenous fluorophore"
#> 4 antibodies_path "extras/antibodies.tsv"
#> 5 contributors_path "extras/contributors.tsv"
#> 6 data_path "."
#> 7 dataset_type "Auto-fluorescence"
#> 8 intended_tile_overlap_percentage ""
#> 9 is_image_preprocessing_required "no"
#> 10 is_targeted "No"
#> 11 metadata_schema_id "c9c6a02b-010e-4217-96dc…
#> 12 preparation_protocol_doi "https://dx.doi.org/10.1…
#> 13 scan_direction ""
#> 14 source_storage_duration_unit "hour"
#> 15 source_storage_duration_value "2"
#> 16 tile_configuration "Not applicable"
#> 17 tiled_image_columns ""
#> 18 tiled_image_count ""
#> 19 time_since_acquisition_instrument_calibration_unit ""
#> 20 time_since_acquisition_instrument_calibration_value ""
sample_metadata("8ecdbdc3e2d04898e2563d666658b6a9")
#> # A tibble: 5 × 2
#> Key Value
#> <chr> <chr>
#> 1 donor.Age "71 years"
#> 2 donor.Apolipoprotein E phenotype "Apolipoprotein E phenotype "
#> 3 donor.Pathology note "Pathology note "
#> 4 donor.Race "White "
#> 5 donor.Sex "Male "
donor_metadata("b2c75c96558c18c9e13ba31629f541b6")
#> # A tibble: 8 × 2
#> Key Value
#> <chr> <chr>
#> 1 Age "41 years"
#> 2 Body Mass Index "37.1 kg/m2"
#> 3 Cause of Death "Cerebrovascular accident "
#> 4 Death Event "Natural causes "
#> 5 Mechanism of Injury "Intracranial hemorrhage "
#> 6 Race "White "
#> 7 Sex "Female "
#> 8 Social History "Smoker "Derived Data
Some datasets from the Dataset entity have derived
(support) dataset(s). Use dataset_derived() to retrieve. A
tibble with selected details will be retrieved as if the given dataset
has a support dataset; otherwise, nothing returns.
# no derived/support dataset
dataset_uuid_1 <- "3acdb3ed962b2087fbe325514b098101"
dataset_derived(uuid = dataset_uuid_1)
#> NULL
# has derived/support dataset
dataset_uuid_2 <- "baf976734dd652208d13134bc5c4594b"
dataset_derived(uuid = dataset_uuid_2) |> glimpse()
#> Rows: 1
#> Columns: 6
#> $ uuid <chr> "bbbf5a5b29986dd57910daab00193f35"
#> $ hubmap_id <chr> ""
#> $ data_types <chr> ""
#> $ dataset_type <chr> "Histology [Image Pyramid]"
#> $ status <chr> ""
#> $ last_modified_timestamp <chr> "NA"Sample and Donor have derived
samples and datasets. In the HuBAMPR package,
sample_derived() and donor_derived() functions
are available to use to see the derived datasets and samples from one
sample given sample UUID or one donor given donor UUID. Specify
entity_type parameter to retrieve derived
Dataset or Sample.
sample_uuid <- samples_df |>
filter(last_modified_timestamp >= "2023-01-01" &
last_modified_timestamp <= "2023-10-01",
organ == "Kidney (Left)") |>
head(1) |>
pull(uuid)
sample_uuid
#> [1] "c40774aa2f52a2811db15c5ca1949314"
# Derived Datasets
sample_derived(uuid = sample_uuid, entity_type = "Dataset")
#> # A tibble: 12 × 2
#> uuid derived_dataset_count
#> <chr> <int>
#> 1 4fddf6de0f42a7e2648b547affefc234 1
#> 2 b6fd505b8e8e1829a2783570f9f25256 0
#> 3 c3db2027e148e92fecb85e7d6a1fd708 1
#> 4 3a10030d3323e5353cfdc3ada45cad86 0
#> 5 71642e4c4a9cc12f59f3317b4a19adc9 1
#> 6 bd42ab2f422e45ce6b0f3f55171de8aa 0
#> 7 c8ad223f01b45b25e0dcb07c48a42762 1
#> 8 f7b49444b974c98c6300e0bfe5fc3a75 0
#> 9 beb1b65624fe85b527ee2ce80ef208b2 1
#> 10 c25d6febe5b007ad32bc59246c99833d 0
#> 11 744647801573d1d5700ee7523089734c 1
#> 12 4a98c43ab3b20b06c11dfbed5fd9034b 0
# Derived Samples
sample_derived(uuid = sample_uuid, entity_type = "Sample")
#> # A tibble: 3 × 2
#> uuid organ
#> <chr> <chr>
#> 1 ec54b7d4ab4545166a0d121b3dc1ec3f Kidney (Left)
#> 2 ae98f6ca4f1f9950f7e7e1dedc2acc10 Kidney (Left)
#> 3 b099a37195f532e4b384020dc0e94bb5 Kidney (Left)donor_derived() works same as above.
Provenance Data
For individual entries from Dataset and
Sample entities, uuid_provenance() helps
to retrieve the provenance of the entry as a list of characters (UUID,
HuBMAP ID, and entity type) from the most recent ancestor to the
furthest ancestor. There is no ancestor for Donor UUID, and an empty
list will be returned.
# dataset provenance
dataset_uuid <- "3e4c568d9ce8df9d73b8cddcf8d0fec3"
uuid_provenance(dataset_uuid)
#> [[1]]
#> [1] "eba120ab7bbd864a6f6f3ad41e598d25, Sample"
#>
#> [[2]]
#> [1] "468d73d28b9e8c43ffa5fbd56d8e46e3, Sample"
#>
#> [[3]]
#> [1] "1c749716d32310351cb9557c7e2937a0, Sample"
#>
#> [[4]]
#> [1] "c09f875545a64694d70a28091ffbcf8b, Donor"
# sample provenance
sample_uuid <- "35e16f13caab262f446836f63cf4ad42"
uuid_provenance(sample_uuid)
#> [[1]]
#> [1] "0b43d8d0dbbc5e3923a8b963650ab8e3, Sample"
#>
#> [[2]]
#> [1] "eed96170f42554db84d97d1652bb23ef, Sample"
#>
#> [[3]]
#> [1] "1628b6f7eb615862322d6274a6bc9fa0, Donor"
# donor provenance
donor_uuid <- "0abacde2443881351ff6e9930a706c83"
uuid_provenance(donor_uuid)
#> list()Related Data
Each Collection has related datasets, and use
collection_data() to retrieve.
collections_df <- collections()
collection_uuid <- collections_df |>
filter(last_modified_timestamp <= "2023-01-01") |>
head(1) |>
pull(uuid)
collection_data(collection_uuid)
#> # A tibble: 209 × 7
#> uuid hubmap_id dataset_type_additio…¹ dataset_type last_modified_timest…²
#> <chr> <chr> <chr> <chr> <chr>
#> 1 11b4f41… HBM945.F… CODEX CODEX 2022-09-08
#> 2 c0f9540… HBM363.C… CODEX CODEX 2023-06-16
#> 3 133bc98… HBM893.M… CODEX CODEX 2022-09-08
#> 4 d71d398… HBM673.Z… snATACseq ATACseq 2023-04-20
#> 5 50f4662… HBM395.N… CODEX CODEX 2023-06-16
#> 6 f49b28c… HBM522.L… snATACseq ATACseq 2023-03-06
#> 7 d03a695… HBM654.V… snATACseq ATACseq 2023-04-20
#> 8 a80f8f6… HBM296.N… snATACseq ATACseq 2023-04-20
#> 9 bd4f1b9… HBM376.R… snATACseq ATACseq 2023-03-06
#> 10 1afbbda… HBM693.B… snATACseq ATACseq 2023-04-20
#> # ℹ 199 more rows
#> # ℹ abbreviated names: ¹dataset_type_additional_information,
#> # ²last_modified_timestamp
#> # ℹ 2 more variables: status <chr>, organ <chr>Each publication has related datasets, samples, and donors, and use
publication_data() to see, while specifying
entity_type parameter to retrieve derived
Dataset or Sample.
publications_df <- publications()
publication_uuid <- publications_df |>
filter(publication_venue == "Nature") |>
head(1) |>
pull(uuid)
publication_data(publication_uuid, entity_type = "Dataset")
#> # A tibble: 209 × 2
#> dataset_type uuid
#> <chr> <chr>
#> 1 CODEX 9e28cdbdb5bc5b3cf7299f13635eebc9
#> 2 CODEX e6fd525b837f4cf736c8af830f4f750f
#> 3 RNAseq c591e842e21e4aae360cba2b45deba4b
#> 4 RNAseq 3b3f5daa672e3cd0106b368a43ff3354
#> 5 ATACseq 77f80b6c8ed9f05884edcd55e6885246
#> 6 RNAseq d611a7de3a07bd5b88e669ece6a8c9b0
#> 7 RNAseq 4c26f91beabafb3290fad2bfbdc68ab3
#> 8 RNAseq 92ad89cb6b937a8af1778dbe985da7b1
#> 9 RNAseq af118b12493d5d3794b38b30c3aced45
#> 10 ATACseq e2d35f4ec17d63e11f2ce7426b0c0258
#> # ℹ 199 more rows
publication_data(publication_uuid, entity_type = "Sample")
#> # A tibble: 163 × 2
#> dataset_type uuid
#> <chr> <chr>
#> 1 Sample 2a70c0c2648986e08b8c5b81e6561f68
#> 2 Sample 4befc15642433858a6bc34b69596e160
#> 3 Sample 3b46c8d8fc519586fe1c9798ac95b245
#> 4 Sample 855045d4390dfa55cb95435eb9c3bcd0
#> 5 Sample 1de4ad9bd516660043b8e190e8978045
#> 6 Sample cb53dd247ec7e651161932fde83b4ca8
#> 7 Sample ac851bd82b0745e9c39fd3c79e4b8085
#> 8 Sample f7b37eff8a36eb3852a373cc4878f50e
#> 9 Sample 5d079766bf17544745c38d88d6923dbf
#> 10 Sample 3135de0b9f359f09f9e6891784b88cae
#> # ℹ 153 more rowsAdditional Information
To read the textual description of one Collection or
Publication, use collection_information()
or publication_information() respectively.
collection_information(uuid = collection_uuid)
#> Title
#> High Resolution Single Cell Maps Reveals Distinct Cell Organization and Function Across Different Regions of the Human Intestine
#> Description
#> The colon is a complex organ that promotes digestion, extracts nutrients, participates in immune surveillance, maintains critical symbiotic relationships with microbiota, and affects overall health. To better understand its organization, functions, and its regulation at a single cell level, we performed CODEX multiplexed imaging, as well as single nuclear RNA and open chromatin assays across eight different intestinal sites of four donors. Through systematic analyses we find cell compositions differ dramatically across regions of the intestine, demonstrate the complexity of epithelial subtypes, and find that the same cell types are organized into distinct neighborhoods and communities highlighting distinct immunological niches present in the intestine. We also map gene regulatory differences in these cells suggestive of a regulatory differentiation cascade, and associate intestinal disease heritability with specific cell types. These results describe the complexity of the cell composition, regulation, and organization for this organ, and serve as an important reference map for understanding human biology and disease.
#> DOI
#> - https://doi.org/10.35079/HBM692.JRZB.356
#> URL
#> - 10.35079/HBM692.JRZB.356
publication_information(uuid = publication_uuid)
#> Title
#> Organization of the human intestine at single-cell resolution
#> Abstract
#> We investigated the spatial arrangement of individual cells using multiplexed imaging, as well as single-nucleus RNA and open chromatin assays, across eight different regions of the intestine from nine donors. Through comprehensive analyses, we observed significant variations in cell compositions among the intestinal regions and the intricate nature of epithelial subtypes. Furthermore, we discovered that similar cell types form distinct neighborhoods and communities, highlighting the presence of unique immunological niches within the intestine. Additionally, we identified gene regulatory differences within these cells, suggesting the existence of a regulatory differentiation cascade, and established associations between specific cell types and the heritability of intestinal diseases. These findings elucidate the intricate cell composition, regulation, and spatial organization within this organ, providing a valuable reference map for advancing our understanding of human biology and disease.
#> Manuscript
#> - Nature: https://doi.org/10.1038/s41586-023-05915-x
#> Corresponding Authors
#> - John Hickey 0000-0001-9961-7673
#> Data Types
#> - CODEX
#> - RNAseq
#> - ATACseq
#> Organs
#> - Large Intestine
#> - Small IntestineSome additional contact/author/contributor information can be
retrieved using dataset_contributor() for
Dataset entity, collection_contact() and
collection_contributors() for Collection
entity, or publication_authors() for
Publication entity.
# Dataset
dataset_contributors(uuid = dataset_uuid)
#> # A tibble: 2 × 11
#> affiliation display_name email first_name is_contact is_operator
#> <chr> <chr> <chr> <chr> <chr> <chr>
#> 1 "University of Californi… Xingzhao Wen xzwe… Xingzhao Yes Yes
#> 2 "University of Californi… Sheng Zhong szho… Sheng Yes No
#> # ℹ 5 more variables: is_principal_investigator <chr>, last_name <chr>,
#> # metadata_schema_id <chr>, middle_name_or_initial <chr>, orcid <chr>
# Collection
collection_contacts(uuid = collection_uuid)
#> # A tibble: 2 × 3
#> name affiliation orcid_id
#> <chr> <chr> <chr>
#> 1 Michael P. Snyder Stanford University 0000-0003-0784-7987
#> 2 John W. Hickey Stanford University 0000-0001-9961-7673
collection_contributors(uuid = collection_uuid)
#> # A tibble: 33 × 3
#> name affiliation orcid_id
#> <chr> <chr> <chr>
#> 1 John W. Hickey Stanford University 0000-0001-9961-7673
#> 2 Winston R. Becker Stanford University 0000-0001-7876-5060
#> 3 Stephanie A. Nevins Stanford University 0000-0002-2244-6703
#> 4 Aaron Horning Stanford University 0000-0003-3247-0798
#> 5 Almudena Espin Perez Stanford University 0000-0002-8329-9196
#> 6 Chenchen Zhu Stanford University 0000-0003-2165-9456
#> 7 Bokai Zhu Stanford University 0000-0003-3599-9419
#> 8 Bei Wei Stanford University 0000-0003-1516-8802
#> 9 Roxanne Chiu Stanford University 0000-0002-7306-9186
#> 10 Derek C. Chen Stanford University 0000-0001-9652-4885
#> # ℹ 23 more rows
# Publication
publication_authors(uuid = publication_uuid)
#> # A tibble: 3 × 3
#> name affiliation orcid_id
#> <chr> <chr> <chr>
#> 1 John Hickey Stanford University 0000-0001-9961-7673
#> 2 Chiara Caraccio Stanford University 0000-0002-3580-1348
#> 3 Garry Nolan Stanford University 0000-0002-8862-9043File Transfer
For each dataset, there are corresponding data files. Most of the datasets’ files are available on HuBMAP Globus with corresponding URLs. Some of the datasets’ files are not available via Globus but can be accessed via dbGAP (database of Genotypes and Phenotypes) and/or SRA (Sequence Read Archive). However, some of the datasets’ files are not available on any authorized platform.
Each dataset available on Globus has different components of data-related files to preview and download, including but not limited to images, metadata files, downstream analysis reports, raw data products, etc.
Use bulk_data_transfer() to know whether data files are
open-accessed or restricted. Only open-accessed files can be downloaded
for downstream analysis.
Files are publicly accessible
HuBMAP stored all public data files on Globus, which is an
open-source and safe platform for large-size data storage. For every
dataset in which the data files can be publicly accessed, the
bulk_data_transfer() function will direct to the
corresponding Globus webpage in Chrome.
uuid_globus <- "d1dcab2df80590d8cd8770948abaf976"
bulk_data_transfer(uuid_globus)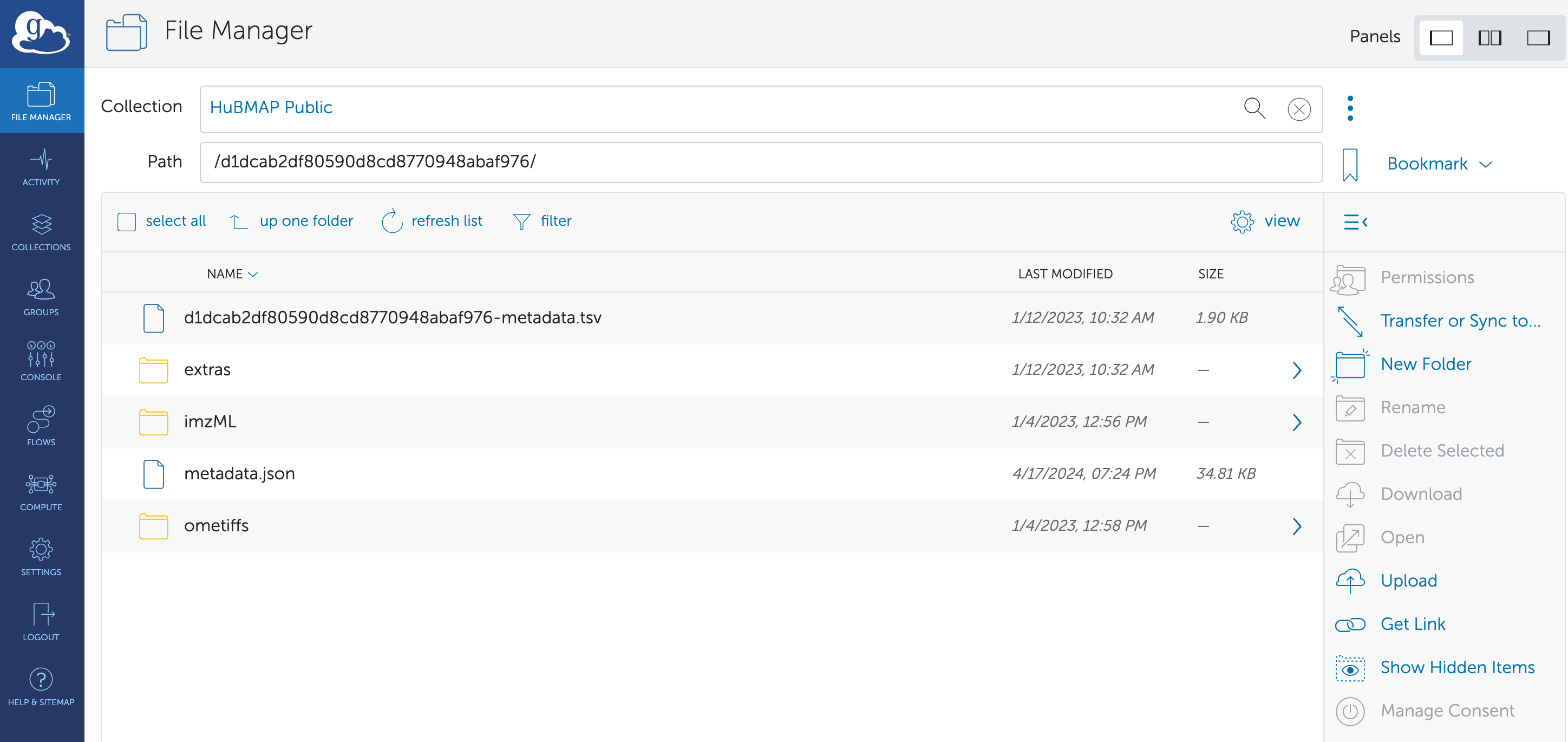
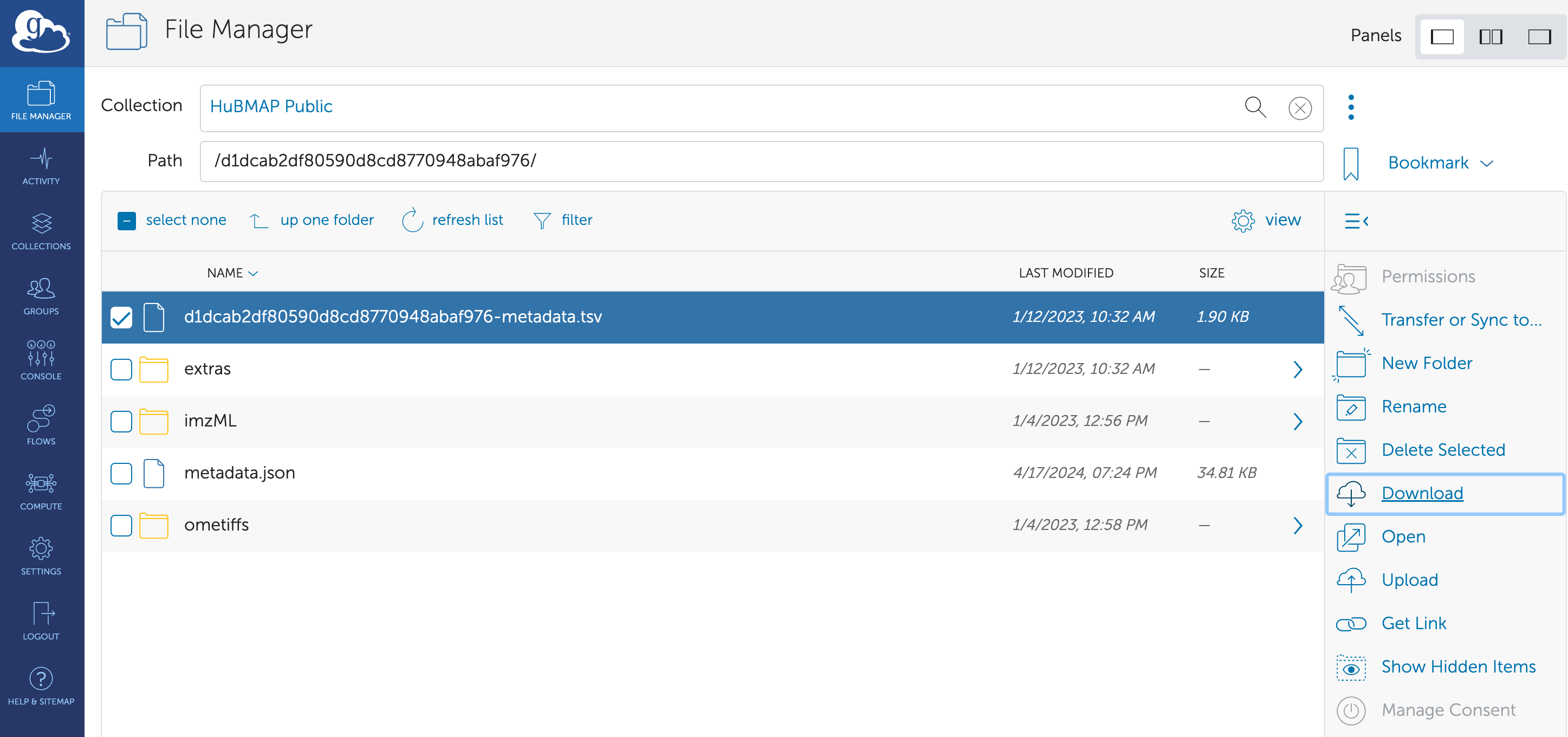
By selecting the data file and clicking on the “Download” button, the data file can be downloaded to the specific directory.
Alternative data transfer method using rglobus package
Martin Morgan, one of the HuBMAPR package creators,
generated an experimental package called rglobus.
Globus is in part a cloud-based file transfer service, available at
https://www.globus.org/. This package provides an
R client with the ability to discover and navigate collections
and to transfer files and directories between collections. Therefore,
rglobus is an alternative method to transfer HuBMAP data
files on the local computer using HuBMAP dataset UUID.
rglobus has the vignette documentation here
using the HuBMAP collection as the main example to illustrate how to
discover and navigate the correct collection and transfer the files.
Since rglobus is an experimental package, the
functionality may not be complete. It is possible to see transfer issues
while using functions. There will be more information updated in the
future. You are welcome to report any issue or provide any comment here to help us
develop.
Files are restricted
For every dataset in which the data files are restricted under dbGAP
or SRA, the bulk_data_transfer() function will print out
the instruction messages. The dbGaP and/or SRA link(s) allow the users
to request the protected-access sequence data from an authenticated
platform.
uuid_dbGAP_SRA <- "d926c41ac08f3c2ba5e61eec83e90b0c"
bulk_data_transfer(uuid_dbGAP_SRA)Pruning cache
Error in bulk_data_transfer(uuid_dbGAP_SRA) :
This dataset contains protected-access human sequence data.
If you are not a Consortium member,
you must access these data through dbGaP if available.
dbGaP authentication is required for downloading.
View documentation on how to attain dbGaP access.
Additional Help: 'https://hubmapconsortium.org/contact-form/'
Navigate to the 'Bioproject' or 'Sequencing Read Archive' links.
dbGaP URL:
https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs002267
Select the 'Run' link on the page to download the dataset.
Additional documentation: https://www.ncbi.nlm.nih.gov/sra/docs/.
SRA URL: https://www.ncbi.nlm.nih.gov/sra/SRX13283313.)Files are unavailable
For every dataset which the data files not available, the
bulk_data_transfer() function will print out the
messages.
uuid_not_avail <- "0eb5e457b4855ce28531bc97147196b6"
bulk_data_transfer(uuid_not_avail)Pruning cache
Error in bulk_data_transfer(uuid_not_avail) :
This dataset contains protected-access human sequence data.
Data isn't yet available through dbGaP,
but will be available soon.
Please contact us via 'https://hubmapconsortium.org/contact-form/'
with any questions regarding this data.
R session information
#> R version 4.5.1 (2025-06-13)
#> Platform: x86_64-pc-linux-gnu
#> Running under: Ubuntu 24.04.2 LTS
#>
#> Matrix products: default
#> BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
#> LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.26.so; LAPACK version 3.12.0
#>
#> locale:
#> [1] LC_CTYPE=C.UTF-8 LC_NUMERIC=C LC_TIME=C.UTF-8 LC_COLLATE=C.UTF-8 LC_MONETARY=C.UTF-8
#> [6] LC_MESSAGES=C.UTF-8 LC_PAPER=C.UTF-8 LC_NAME=C LC_ADDRESS=C LC_TELEPHONE=C
#> [11] LC_MEASUREMENT=C.UTF-8 LC_IDENTIFICATION=C
#>
#> time zone: UTC
#> tzcode source: system (glibc)
#>
#> attached base packages:
#> [1] stats graphics grDevices utils datasets methods base
#>
#> other attached packages:
#> [1] pryr_0.1.6 HuBMAPR_1.2.2 ggplot2_3.5.2 tidyr_1.3.1 dplyr_1.1.4 BiocStyle_2.36.0
#>
#> loaded via a namespace (and not attached):
#> [1] utf8_1.2.6 rappdirs_0.3.3 sass_0.4.10 generics_0.1.4 stringi_1.8.7
#> [6] digest_0.6.37 magrittr_2.0.3 evaluate_1.0.3 grid_4.5.1 RColorBrewer_1.1-3
#> [11] bookdown_0.43 fastmap_1.2.0 lobstr_1.1.2 jsonlite_2.0.0 whisker_0.4.1
#> [16] BiocManager_1.30.26 purrr_1.0.4 scales_1.4.0 codetools_0.2-20 httr2_1.1.2
#> [21] textshaping_1.0.1 jquerylib_0.1.4 cli_3.6.5 rlang_1.1.6 withr_3.0.2
#> [26] cachem_1.1.0 yaml_2.3.10 tools_4.5.1 curl_6.3.0 vctrs_0.6.5
#> [31] rjsoncons_1.3.2 R6_2.6.1 lifecycle_1.0.4 stringr_1.5.1 fs_1.6.6
#> [36] ragg_1.4.0 pkgconfig_2.0.3 desc_1.4.3 pkgdown_2.1.3 pillar_1.10.2
#> [41] bslib_0.9.0 gtable_0.3.6 glue_1.8.0 Rcpp_1.0.14 systemfonts_1.2.3
#> [46] xfun_0.52 tibble_3.3.0 tidyselect_1.2.1 knitr_1.50 farver_2.1.2
#> [51] htmltools_0.5.8.1 labeling_0.4.3 rmarkdown_2.29 compiler_4.5.1 prettyunits_1.2.0